|

|
|
|
|
|
|
 > Proteinanalyse mit dem Massenspektrometer <
> Proteinanalyse mit dem Massenspektrometer <
|
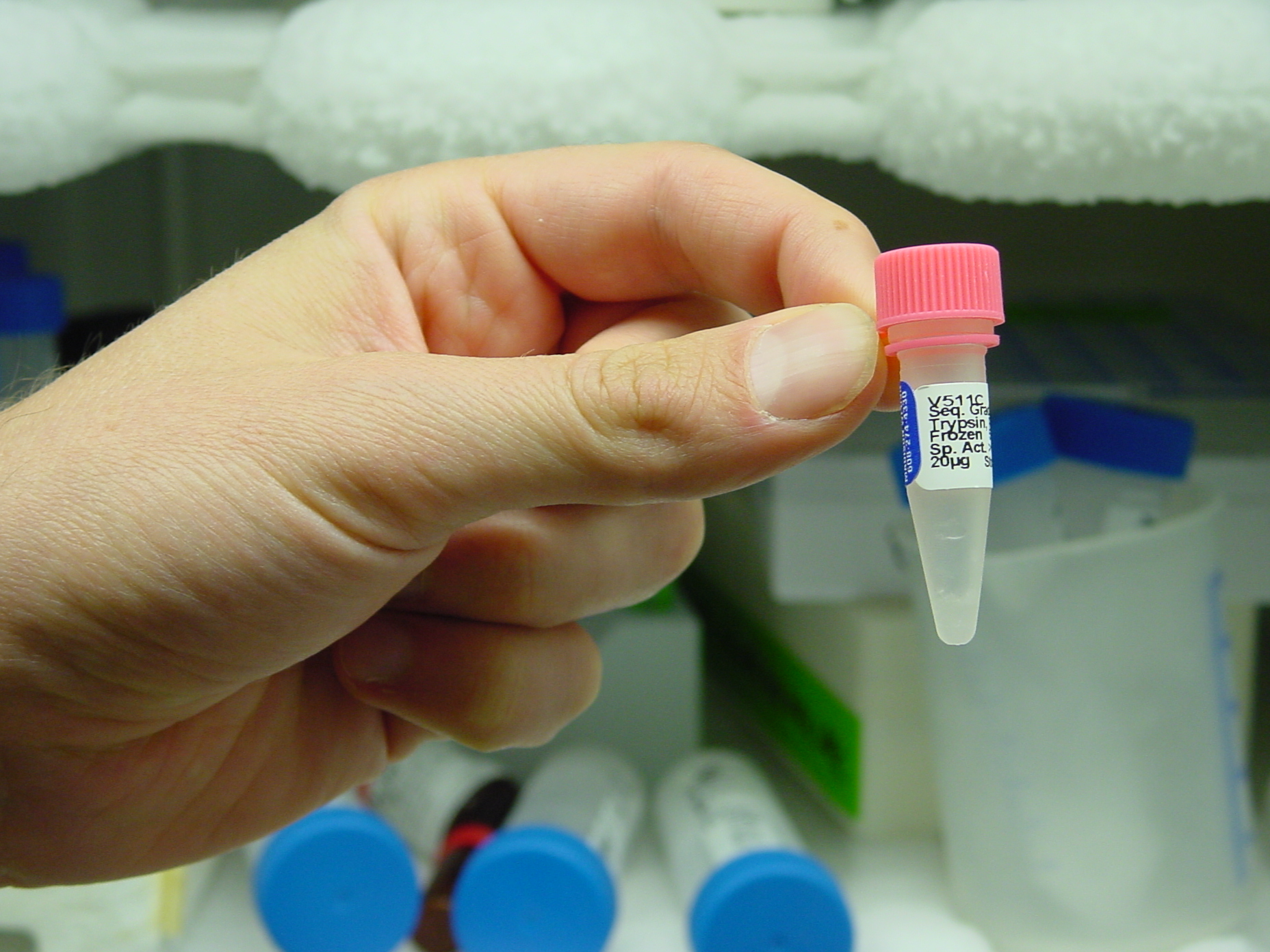
Trypsinlösung gefroren (Lagertemperatur: -80 Grad Celsius)
|
Wie werden Proteine mit einem Massenspektrometer analysiert?
Ein Massenspektrometer kann die Masse von Molekülen exakt bestimmen. Da sich die Masse eines Proteins aus den genau bekannten Massen der einzelnen Aminosäuren zusammensetzt, kann aus der Gesamtmasse auf die mögliche Aminosäurezusammensetzung geschlossen werden. Je grösser die Gesamtmasse ist, desto mehr mögliche Aminosäuresequenzen wird es geben.
Für die Analyse werden Proteine zunächst durch Behandlung mit spezifischen Proteasen in kleinere Fragmente (Peptide) zerlegt. Das wird z.B. mit Trypsin gemacht, einem Verdauungsenzym, das die langen Aminosäureketten der Proteine zerkleinert, in dem es sie an genau definierten Stellen zerschneidet.
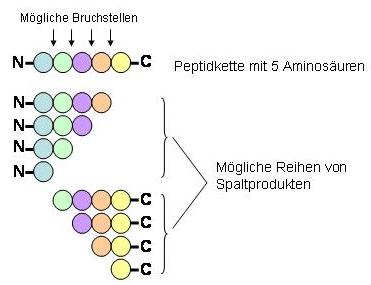
Das Gemisch der einzelnen Peptide wird dann zur Analyse ihrer Aminosäuresequenz in ein Massenspektrometer eingeleitet. Dort werden die Fragmente nach bekannten Regeln noch weiter in Bruchstücke gespalten. Dabei können die Bedingungen so gewählt werden, dass jedes Peptid statistisch nur noch einmal zerfällt, wobei Brüche zwischen allen benachbarten Aminosäuren ähnlich wahrscheinlich sind. Da das Ausgangsmaterial sehr viele Moleküle des jeweiligen Peptids enthält, die statistisch zwischen zwei beliebigen Aminosäuren brechen, ergibt sich eine Population von Peptidfragmenten. Im Prinzip entstehen zwei Reihen von Spaltprodukten, die sich um je eine Aminosäure unterscheiden. Bei der einen Reihe erfolgten die Kettenbrücke des Peptids vom C-Ende her, bei der anderen Reihe vom N-Ende her. Die Massendifferenz zwischen zwei solchen Fragmenten erlaubt die Bestimmung der entsprechenden Aminosäure
In der Praxis ist die Methode nicht so einfach, da sich die beiden Reihen überlagern und ausser den erwarteten Brüchen zwischen den Aminosäuren auch noch zahlreiche andere entstehen können. Proteine können auch chemische Gruppen tragen, die zwar bei der Auswertung berücksichtigt werden können, die aber die Zuordnung der Aminosäuren auf Grund der Massenunterschiede erschwert.
Die Methode eignet sich daher gegenwärtig nur beschränkt zur Analyse von unbekannten Proteinen. Bei Organismen aber, deren Genome sequenziert sind und somit alle möglichen Proteinsequenzen bekannt sind, lässt sich leicht feststellen, ob massenspektrometrisch bestimmte Peptidsequenzen von den Genen des Organismus überhaupt kodiert werden. Umgekehrt kann man aus der Genomsequenz auch ableiten, welche Peptide in einem bestimmten Protein zu erwarten sind und man kann diese Voraussagen in der Realität mit der Massenspektroskopie überprüfen.
Die Genomanalyse (Genomik) ermöglicht es, aufgrund der Gensequenzen vorauszusagen, was für Proteine in einem Organismus auftretenden können. Welche Funktionen diese Proteine haben und wie sie miteinander zusammenspielen wird dadurch aber nicht geklärt. Dies kann näher untersucht werden, indem man ausschliesslich die Proteine, die an einem bestimmten Stoffwechselvorgang beteiligt sind isoliert und mittels Massenspektrometrie und Zuordnung der passenden Genomsequenzen identifiziert.
|
|
|
|